由于我自己也是第一次测试XPS并处理数据,可能理解不会太深,因此本篇内容不会详细地追究细节
PS:本文参考了一系列国内外网站,太多忘记保存了,就不列举了
XPS介绍
XPS全称为X射线光电子能谱,是一种用于测定材料中元素构成、元素化学态和电子态的定量能谱技术,说白了就是用来元素鉴定
XPS的三个主要功能:
确定样品表面10nm厚度内的元素种类(除氢和氦),因为XPS只能检测出原子序数大于等于3的元素
确定元素的相对百分比含量
元素的化学环境(价态等)
通常来说,我们把样品寄出去测试,告诉测试人员要测试的元素,比如C、N、S等,然后等他们测完把数据返还给我们进行分析处理,一般会给你对应的XLS、VGD或VGP文件,后面的两种格式可以直接用Avantage软件打开
XPS的物理原理
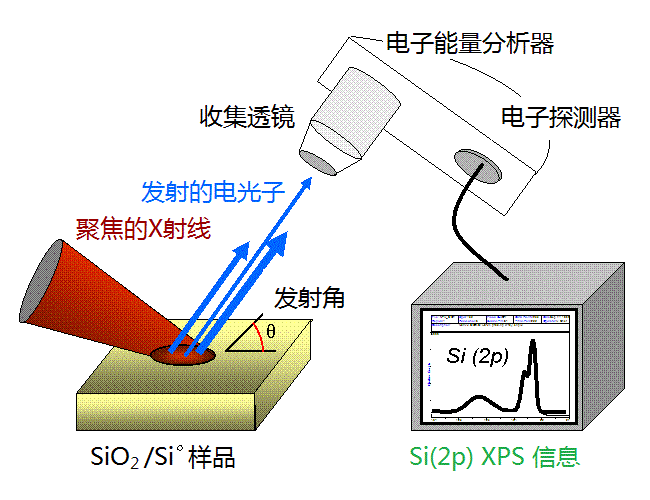
XPS的原理为利用X射线照射样品,激发原子的内层电子及价电子,使其发射出来,激发出来的电子称为光电子。通过测量不同能量的光电子的数目,以结合能(electron binding energy)或光电子的动能(photoelectron kinetic energy)为横坐标,相对强度(counts/s)为纵坐标可做出光电子能谱图
$$E_b(结合能)=hv(光子能量)-E_k(光电子动能)-w(功函数)$$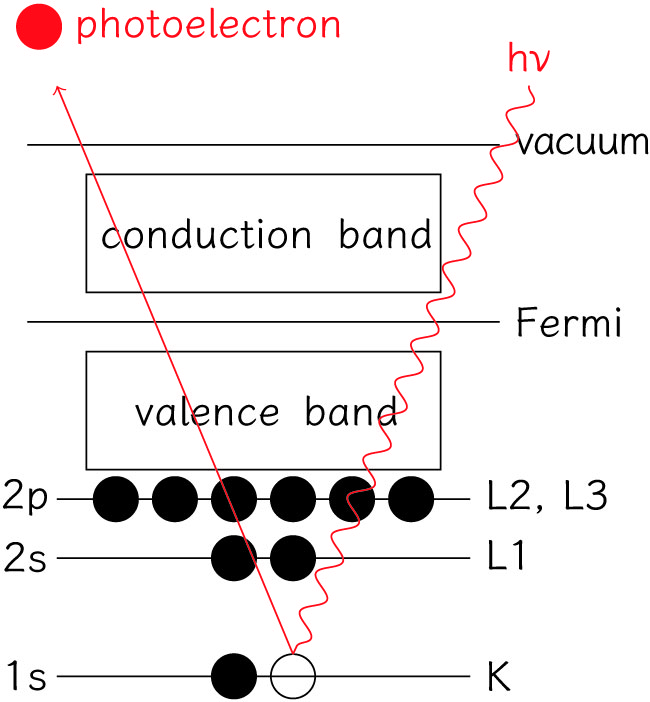
化学态和化学位移
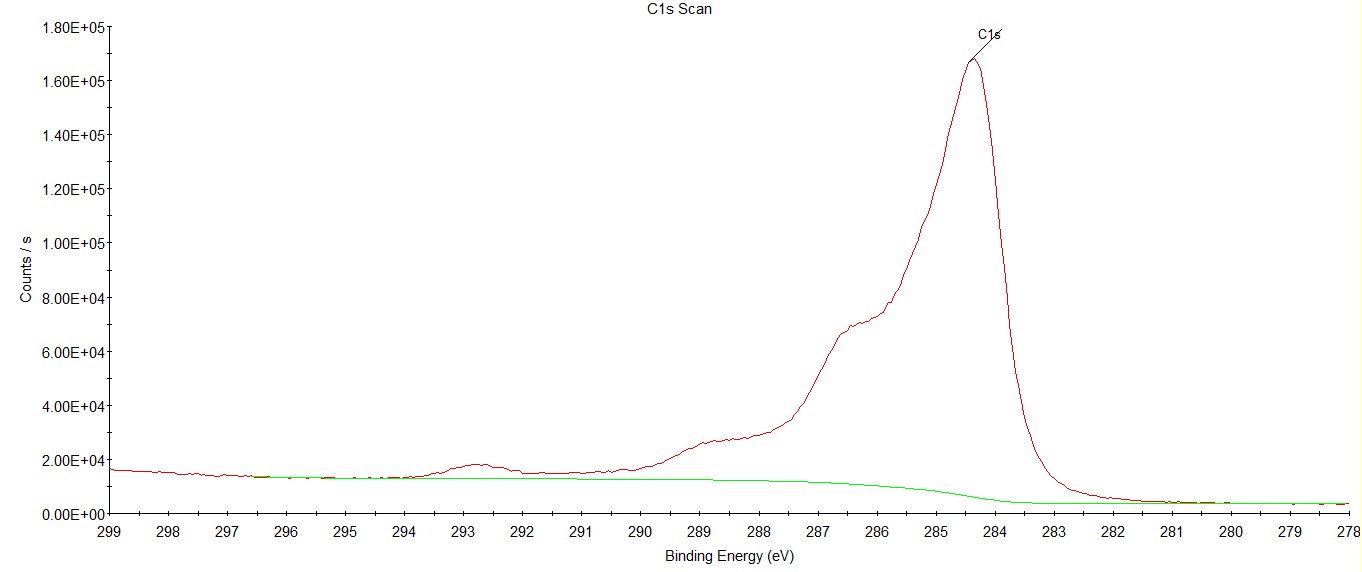
写一下自己的理解:对于同一种元素,以C元素为例,由于样品中C元素所处的化学环境(也就是化学态)不同,在结合能上会产生一些微小的位移(也就是化学位移),这就导致你看到的C元素的峰可能不是一个尖尖的、对称的峰,而可能是不对称,会带点小波浪,甚至是几个挨得很近的峰,这些其实都代表C元素,只不过所处的化学环境不同,所呈现出结合能会有微小的差异,这里所说的化学环境,以C为例可能代表不同的键合状态,比如C-H、C-O或者C=O等,就比如下面这张C1s谱(原始XPS谱,未处理),就能看出实际上有好几个处于不同化学态的C峰
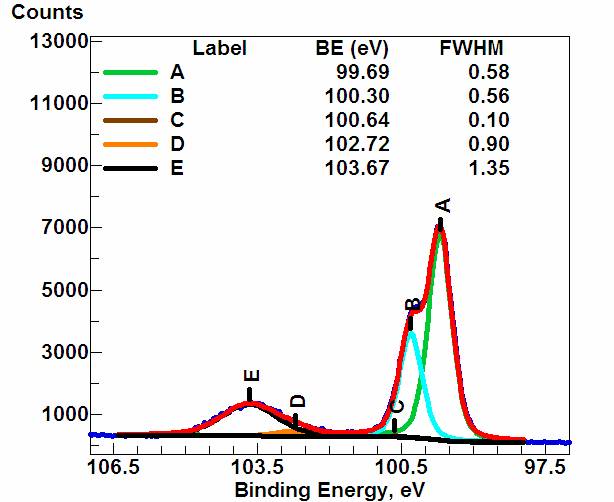
再举一个Si2p的XPS谱,红色的曲线为测得的XPS原始数据,但它实际上是由A、B、C、D、E 五个峰组成,分别对应与Si原子在样品中的五种化学态,其中A、B分别代表两种未被氧化的Si原子,C、D、E分别对应三种不同氧化状态的Si原子
其实,最终我们想要的(放在文章里的)就是上面这种元素谱,也就是要有元素不同化学态对应的峰和拟合后的峰(上图的红线),
XPS全谱
上面只提到了元素谱,其实我看的文章里主要也是用这个,不过你还会发现在给你的数据里除了不同的元素谱还有一张全谱(未处理),大概长下面这个样子:
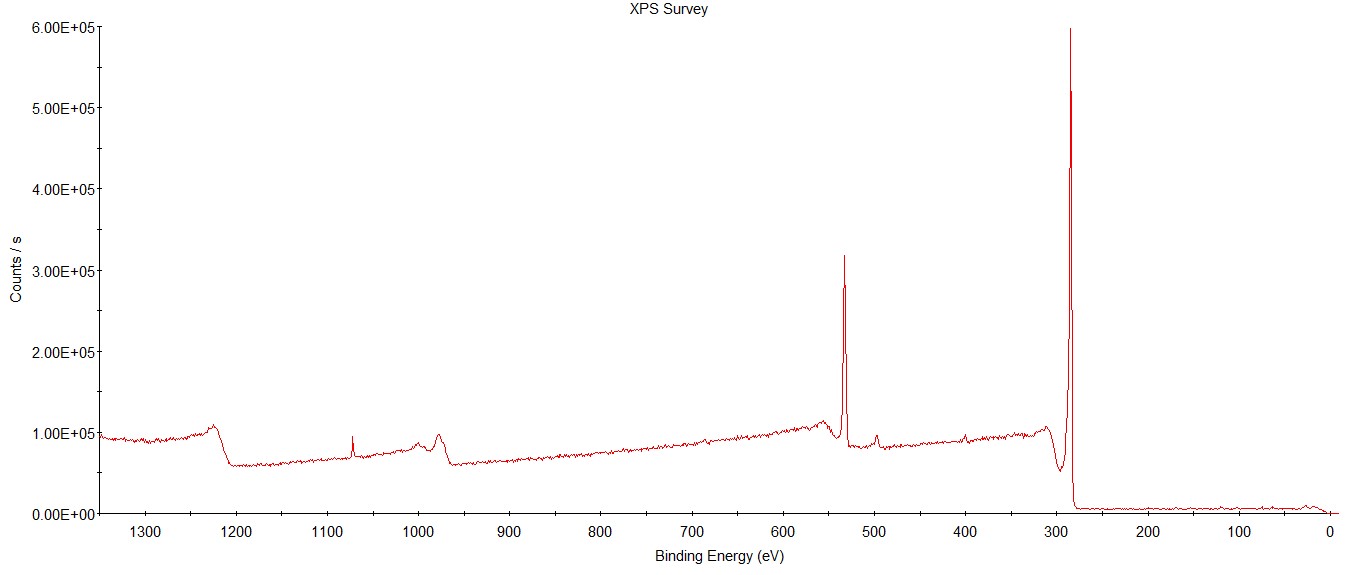
全谱的话,一般可以通过峰的有无来说明是否有该元素,全谱得到的信号会比较粗糙,只是对元素进行了粗略的扫描,所以一般也不会用这个
数据处理
软件:
PS: 也可以通过修改注册表来重置Avantage免费使用时间,下载下面的renew.reg文件并执行即可
下面将使用Avantage软件进行演示
添加背底
首先打开VGP或VGD文件,先看一下有没有背底,没有的话是这个样子
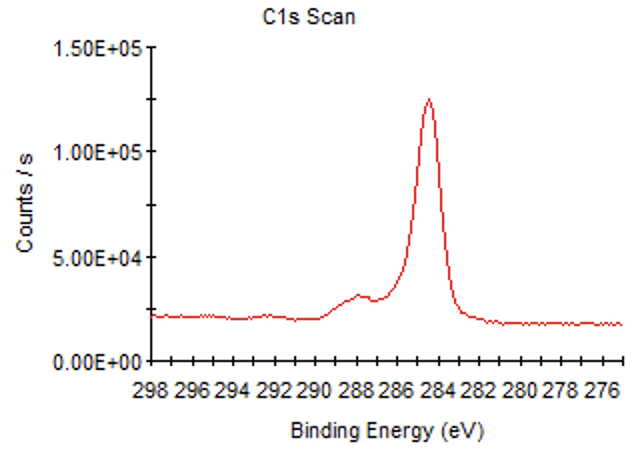
添加后的是这个样子
如果没有添加的话,首先选中任意元素谱图,然后找到峰添加选项
通常直接在smart选项中点击加即可完成添加
也可以手动调整背底的范围
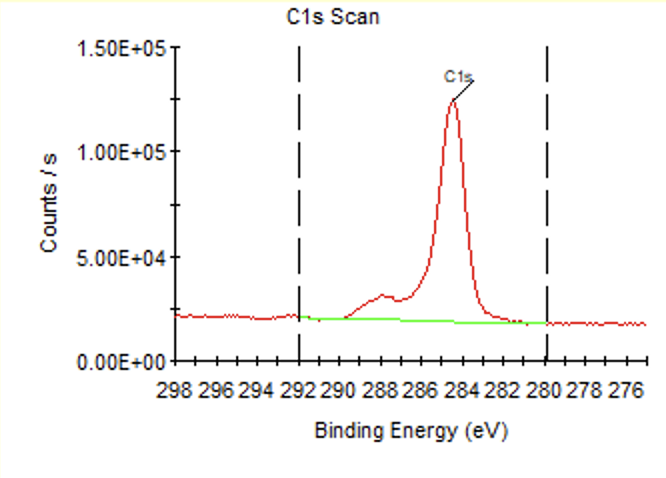
添加背底实际上也会添加一条主峰,如果想删除背底的话直接删除这个峰就好
然后将每个元素谱依次添加背底
填加峰
首先要选中C1s谱,然后从上面的菜单栏找到峰拟合选项打开
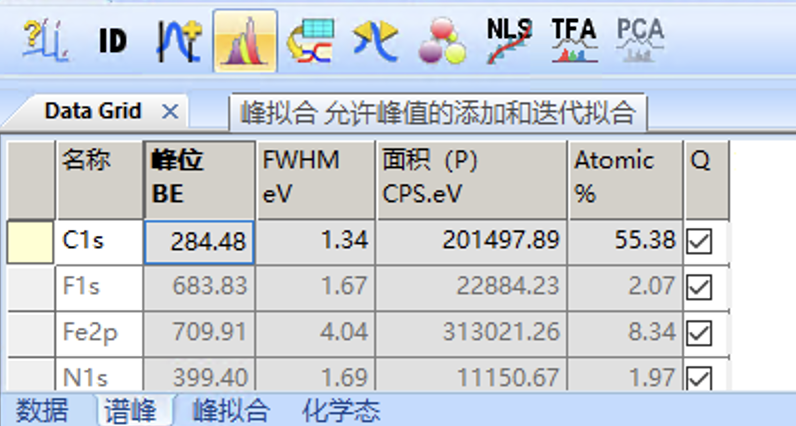
然后先打开Add Fitted Peak选项,准备添加峰(这里要注意,只有s轨道是添加单峰,其他的p、d、f轨道由于都是裂解为双峰)
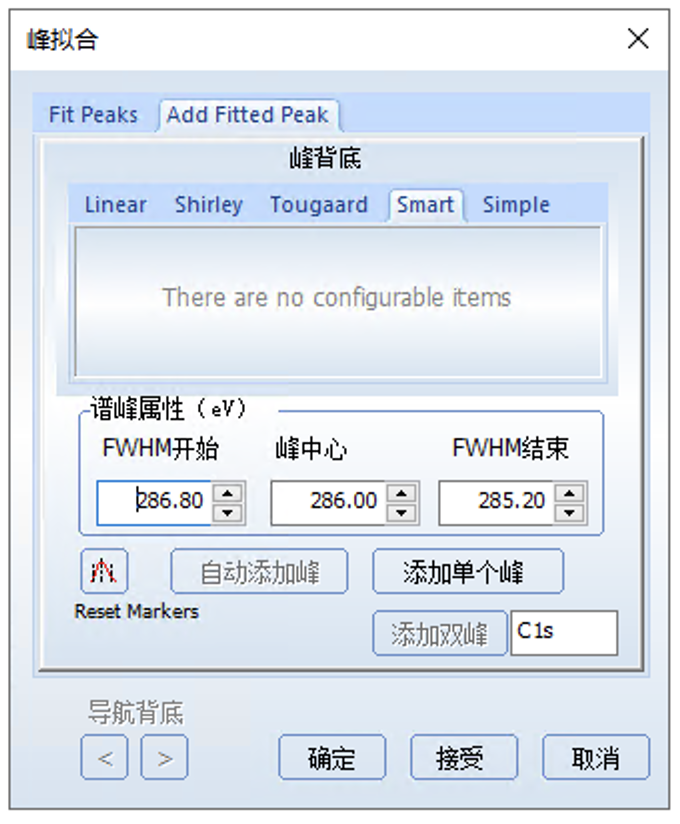
添加峰之前可以在图中标记添加峰的位置,系统会自动标定,但也可以自己调整,然后点击添加单个峰
添加完后就是这个样子,可以根据情况决定是否继续添加
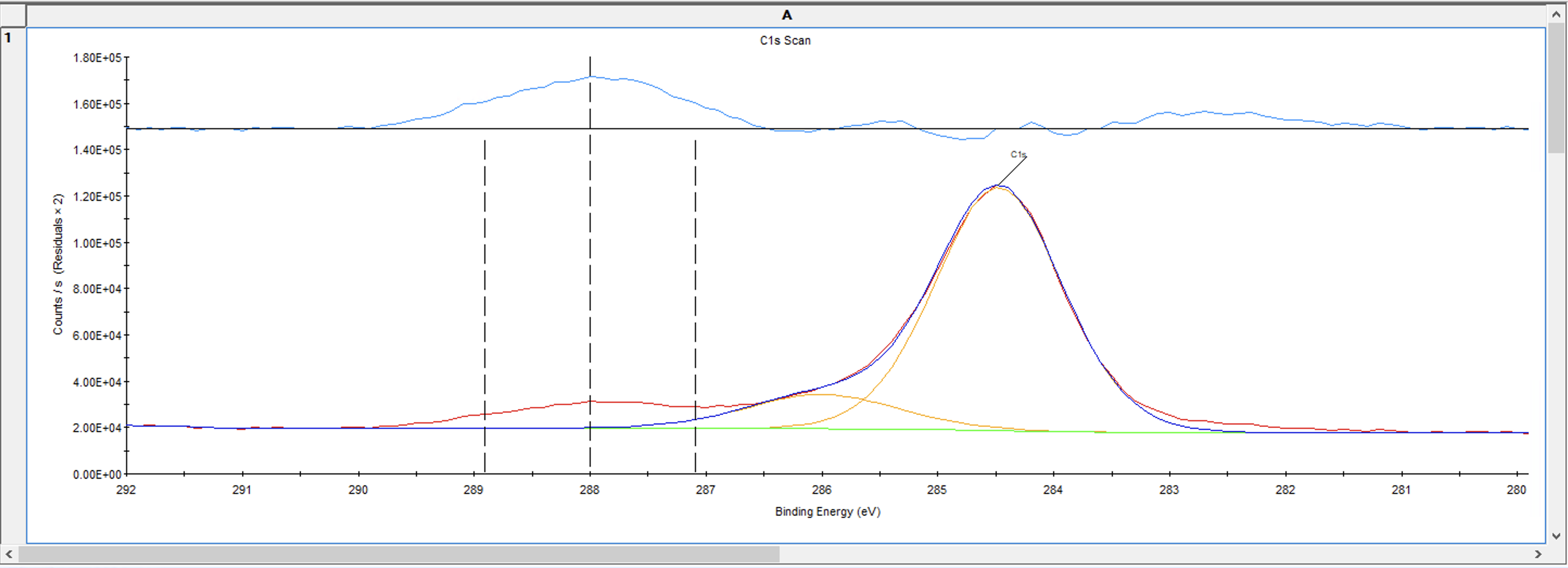
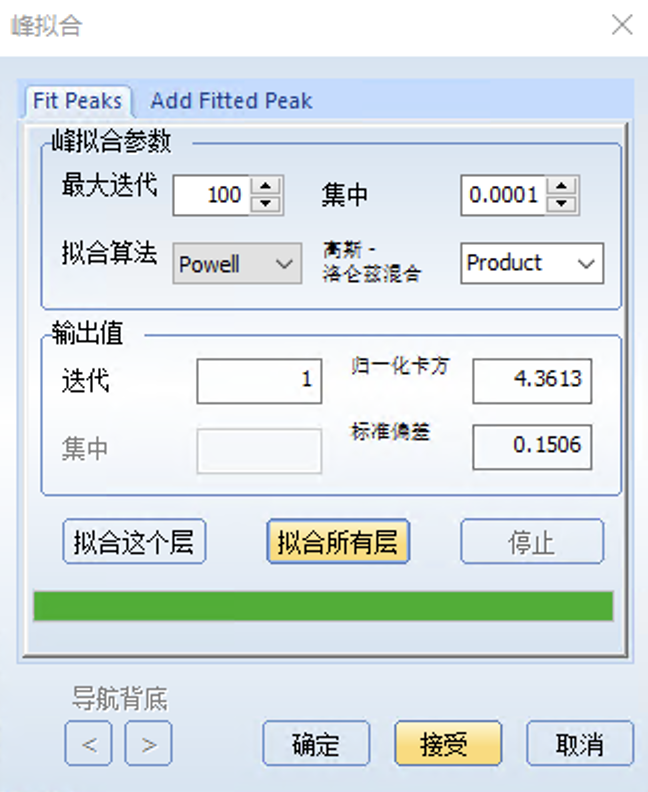
添加完峰后,回到Fit Peaks选项,点击拟合这个层就开始自动拟合,就可以得到拟合结果
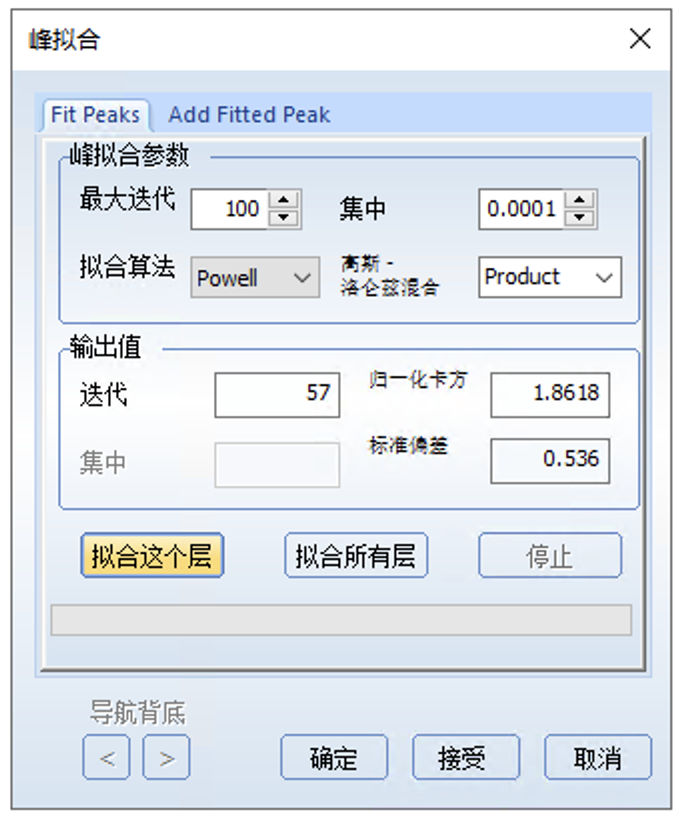
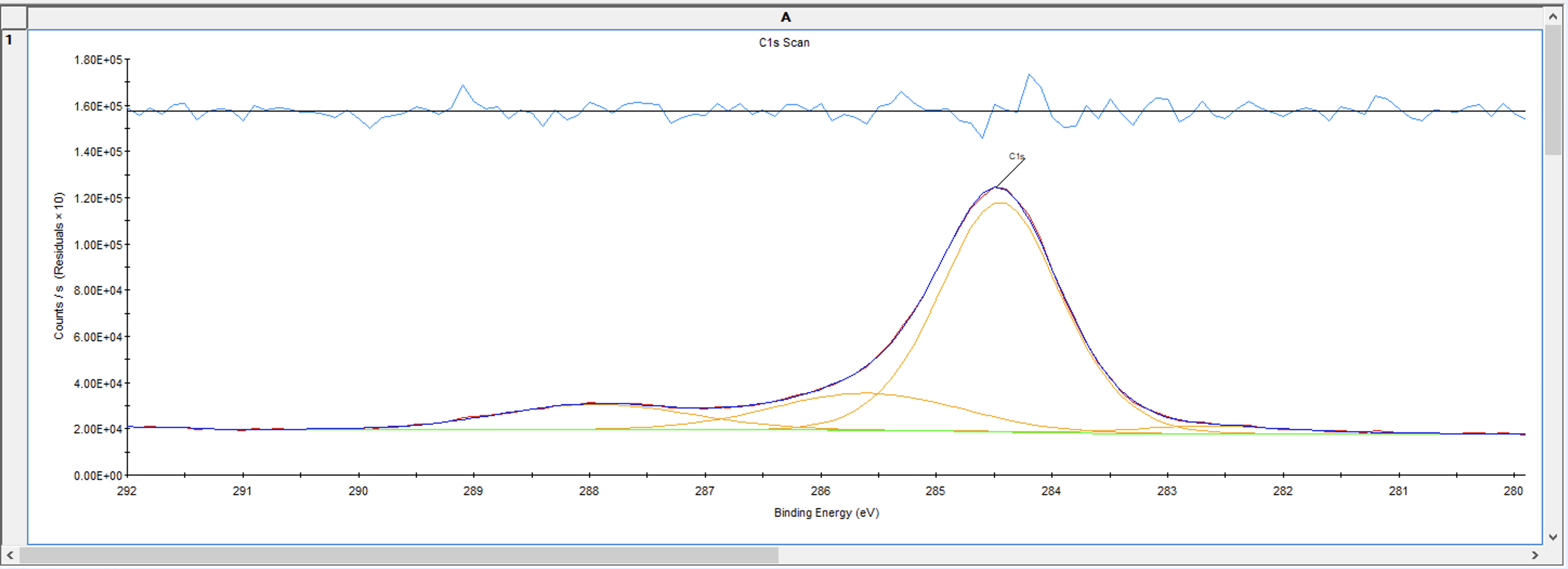
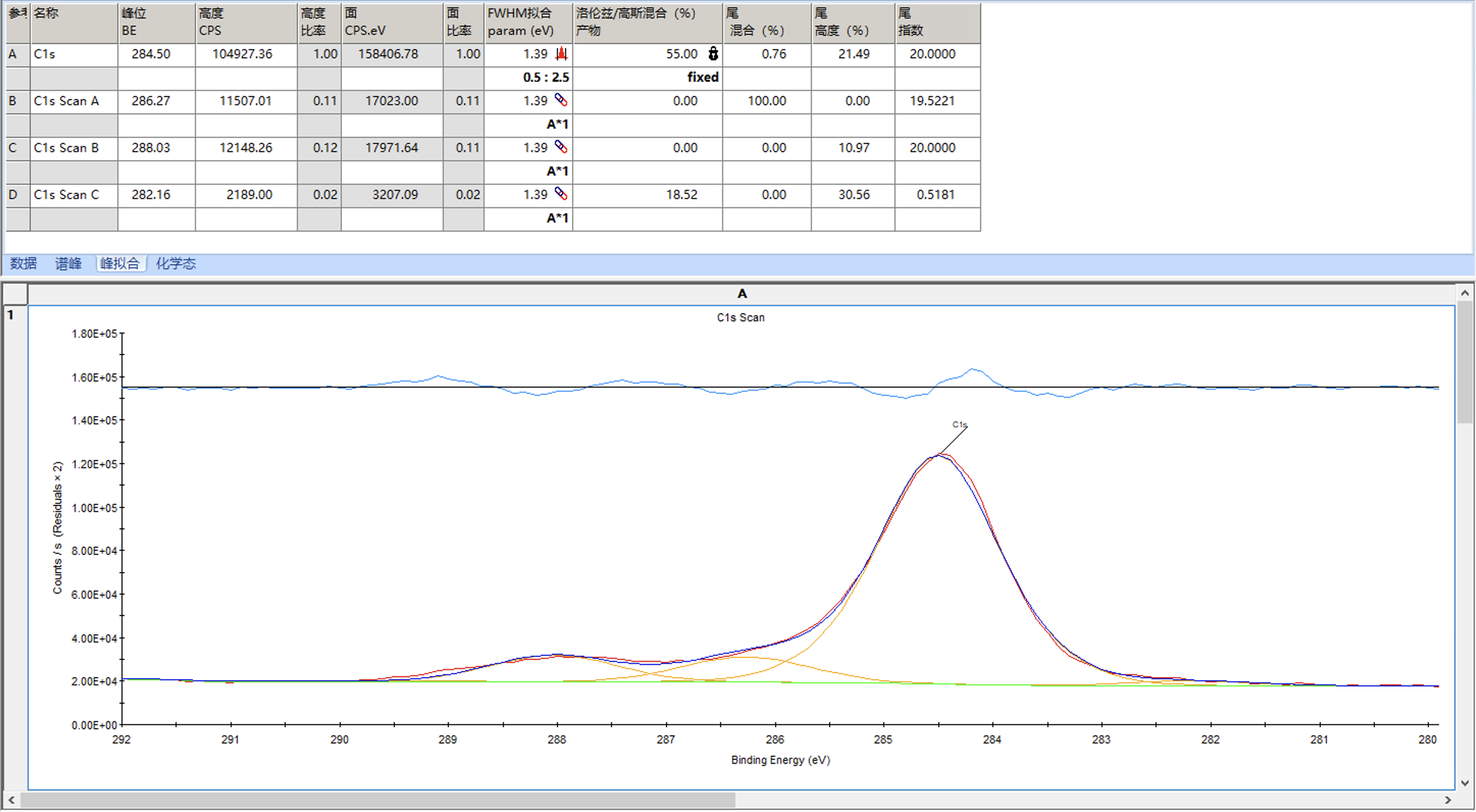
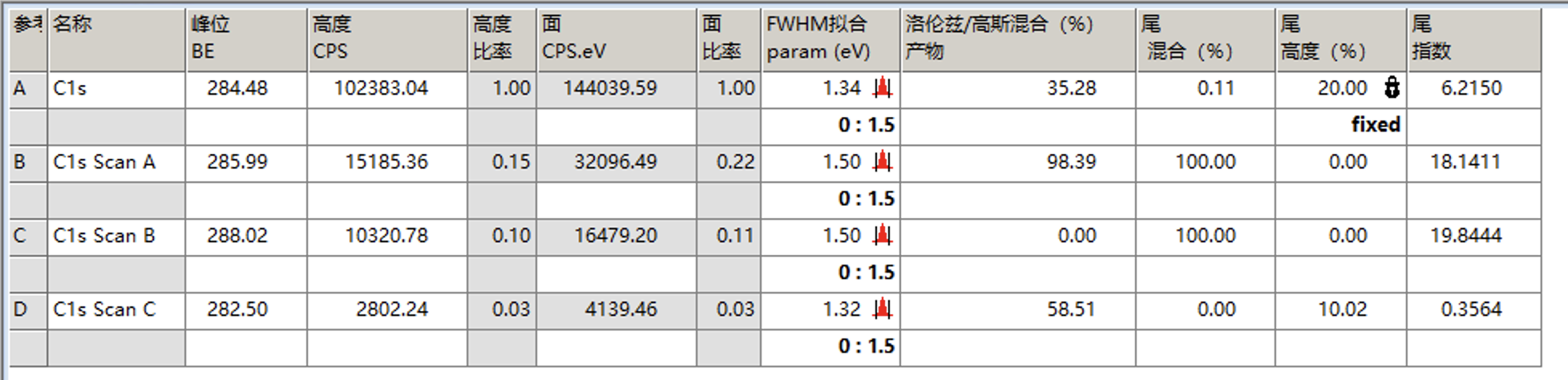
拟合完后,点击下图中的峰拟合,可以看到各个峰的信息,需要关注的是FWHM拟合这一列,代表的是半峰宽,通常不会相差太大,对于C1s来说理论上应该是相同的,但允许有些许偏差,如果各个峰相差较大就需要调整这个值然后重新拟合至半峰宽值接近
例如下面是将A的半峰宽限制在0.5到2.5的范围,然后让B、C、D的半峰宽等于A的半峰宽,然后再次拟合
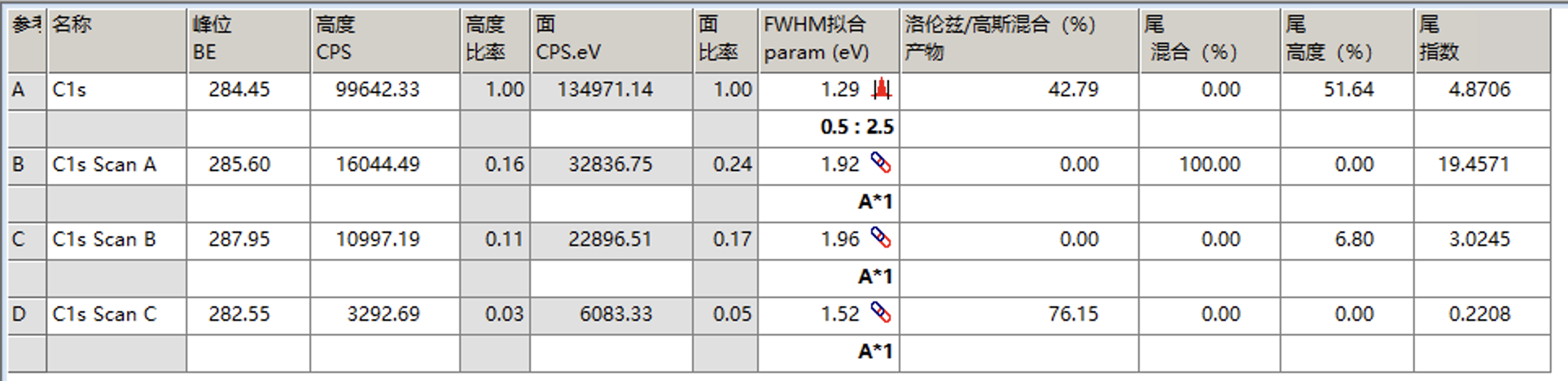
拟合结果如下
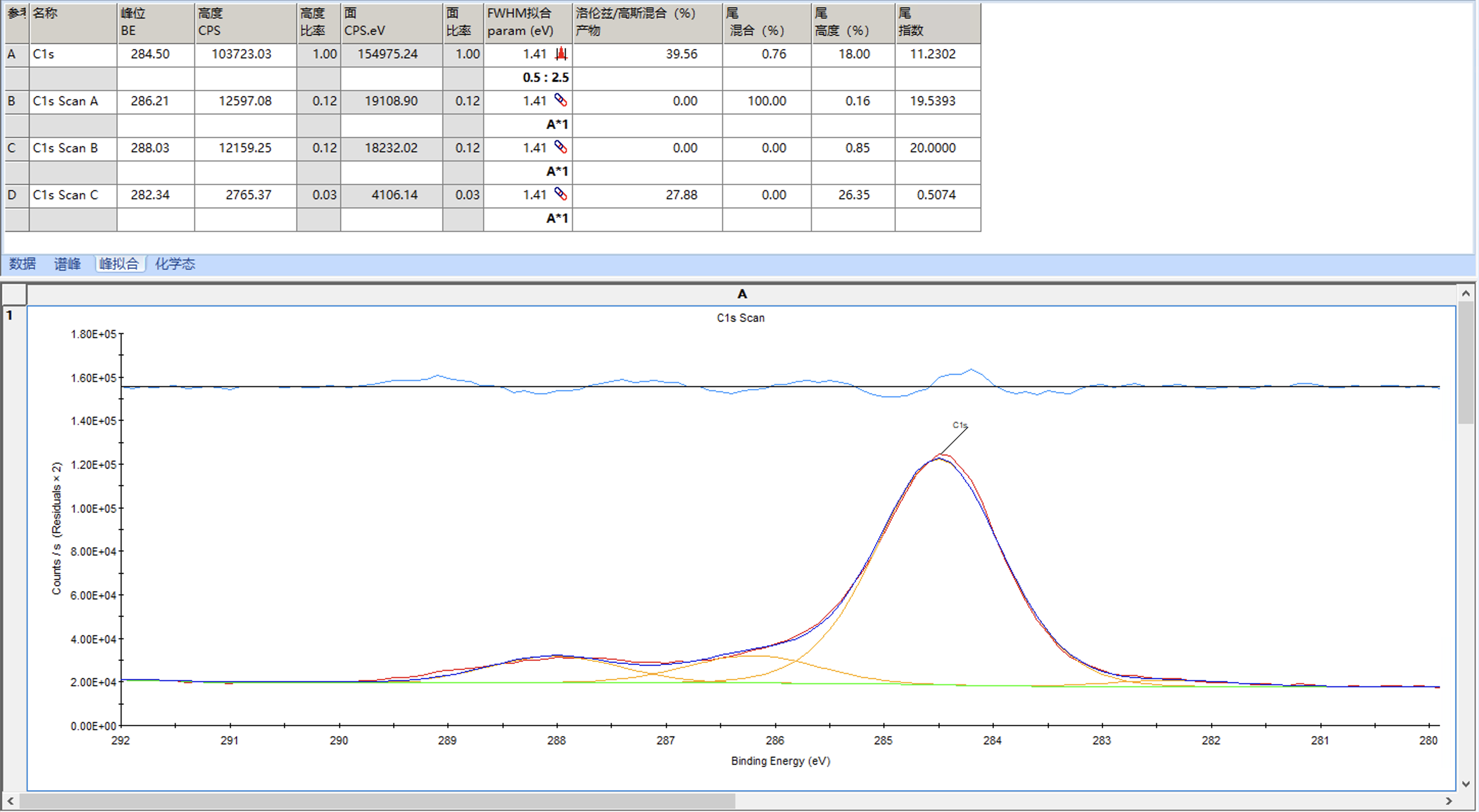
可以发现峰形稍微有点偏差,这时可以考虑调整洛伦兹/高斯比来进行调整,例如这里设置为55并固定
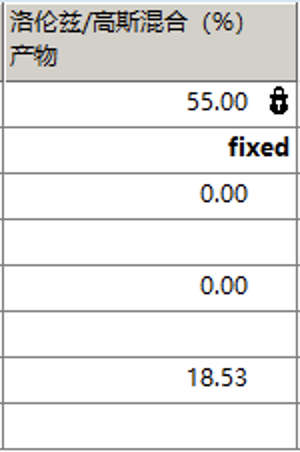
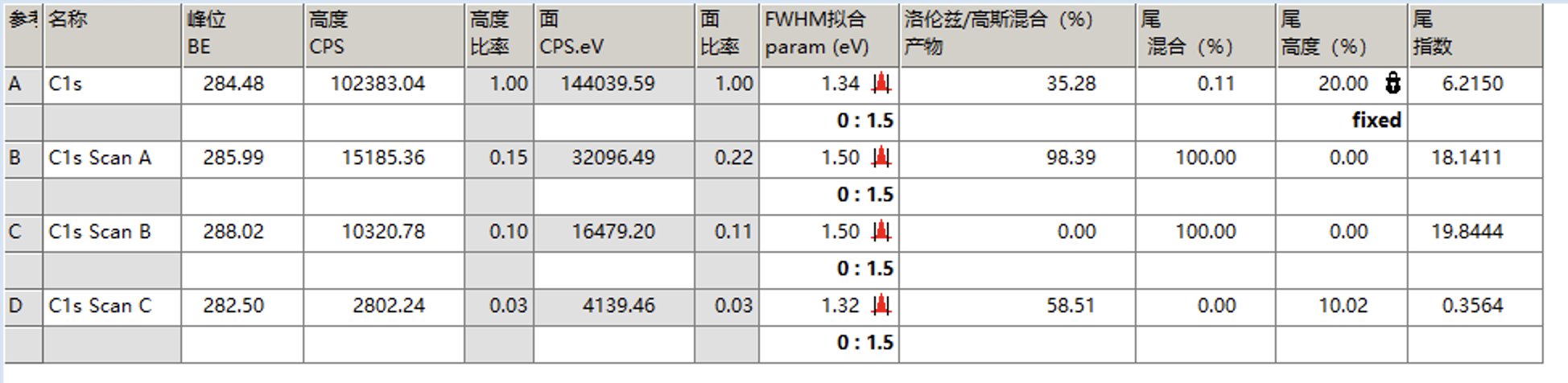
再次拟合后结果如下
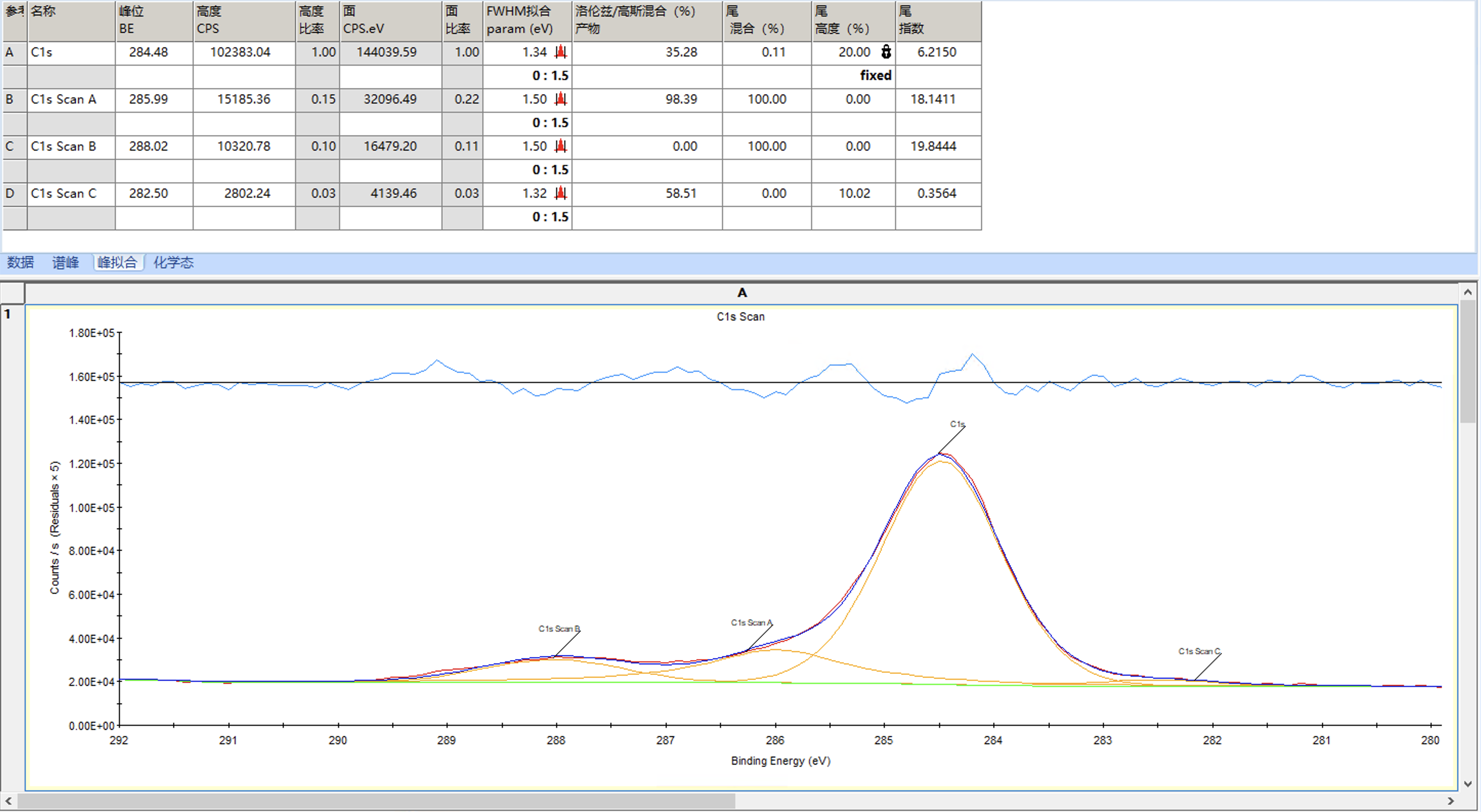
然后我们将所有峰的FWHM拟合的范围改为比当前值略大的范围,取消固定洛伦兹/高斯比,并再次拟合
拟合后结果如下,整体较好
最后点击接受和确定即可
电荷校正
拟合完C1s后,要立刻进行电荷校正,先选中C1s,点击下图中的峰拟合,可以观察刚刚拟合后的结果
接下来要将谱图全选
从上面的菜单点击电荷移位
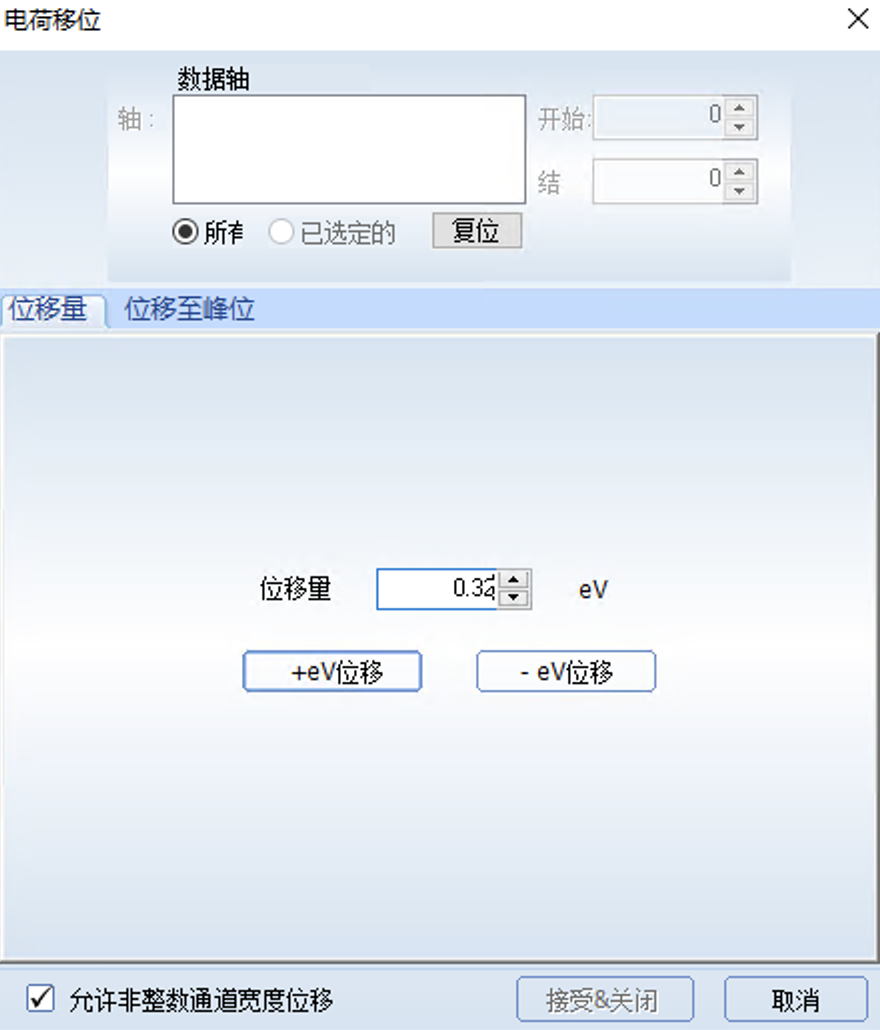
将C1s的峰位值(最接近284.8的峰位)校正为标准峰284.8需要在原来的284.48的基础上加0.32,所以将位移量设为0.32,选中左下角的"允许非整数通道宽度位移",然后点击"+eV位移“就校正完成了
然后我们可以看到C1s已经被校正到284.8了
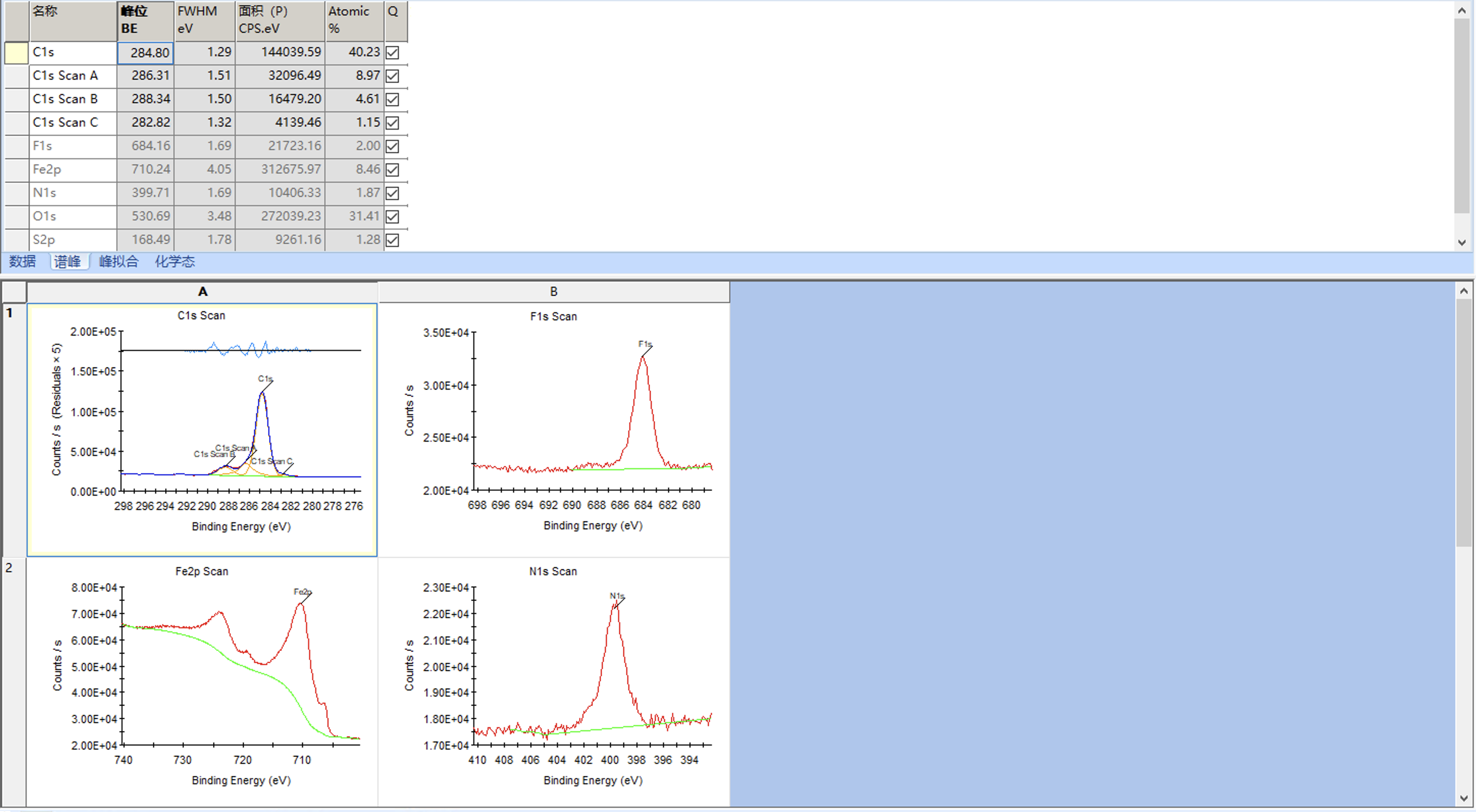
接下来继续将其他元素谱进行加峰拟合,单峰拟合就参考C1s拟合就好
双峰拟合
这里说一下双峰拟合,以Fe2p为例,首先选中Fe2p然后点峰拟合
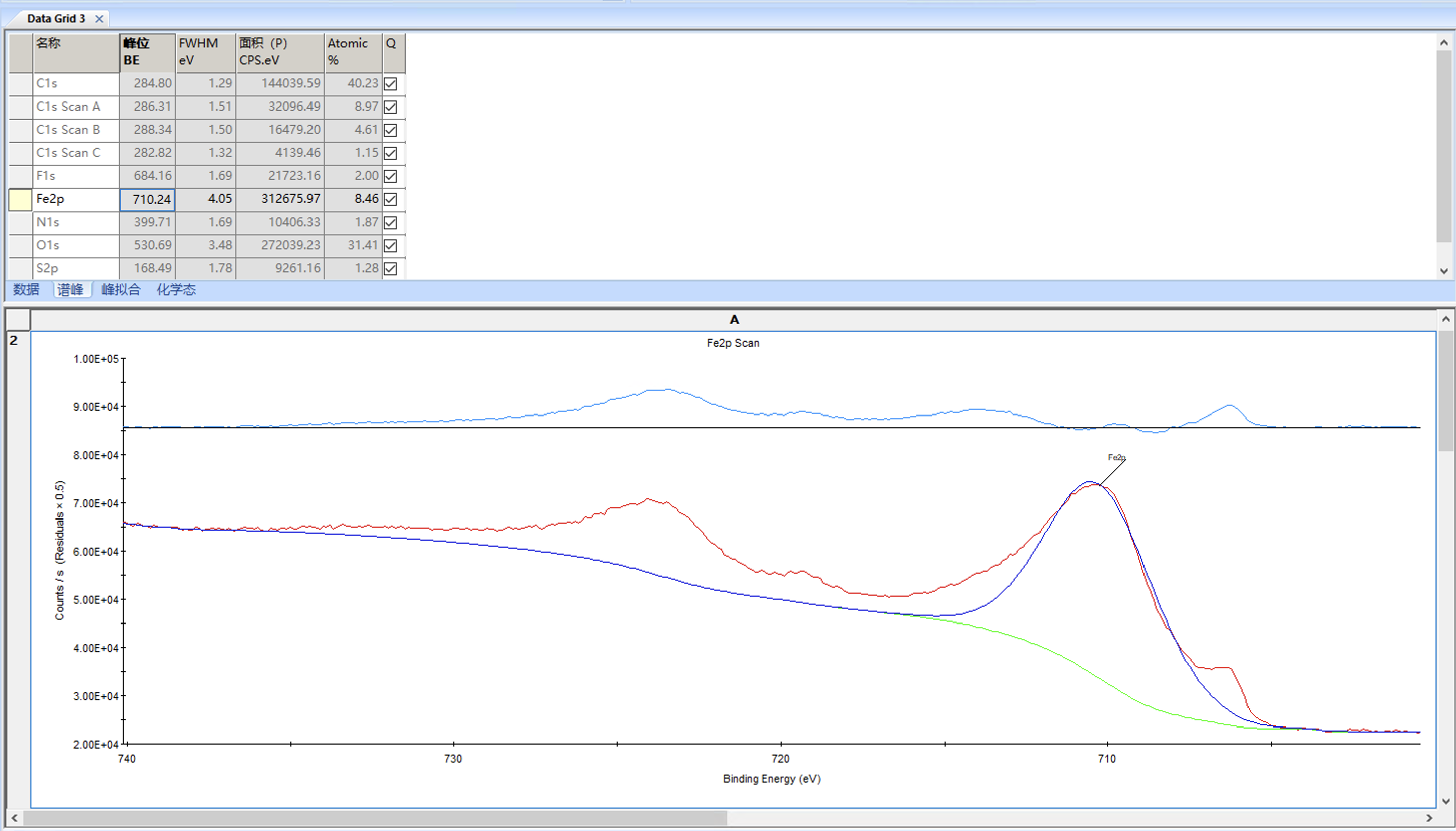
可以看到因为添加了背底所以已经添加了一个峰,但2p轨道裂解为双峰,所以我们添加峰的时候需要先选中这个峰
首先打开峰拟合的Add fitted peak界面
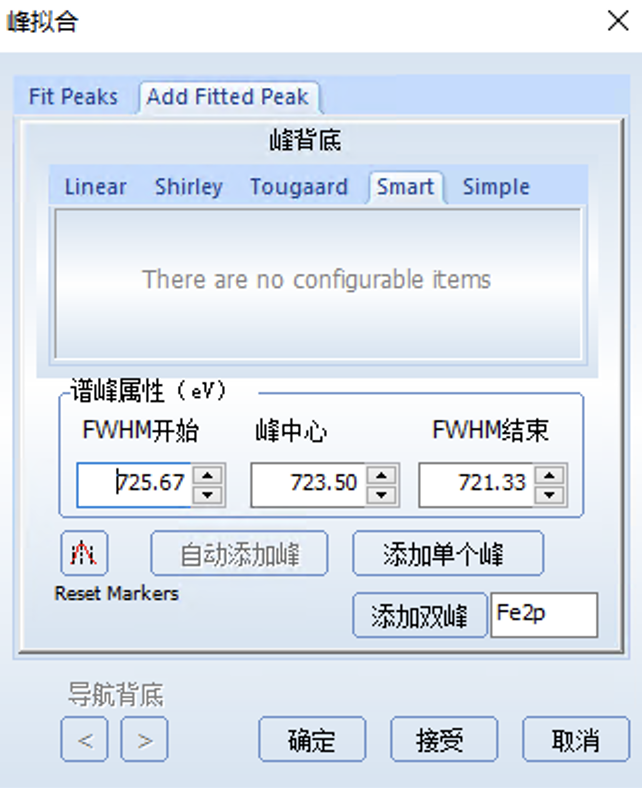
然后选中当前谱图中的这个峰
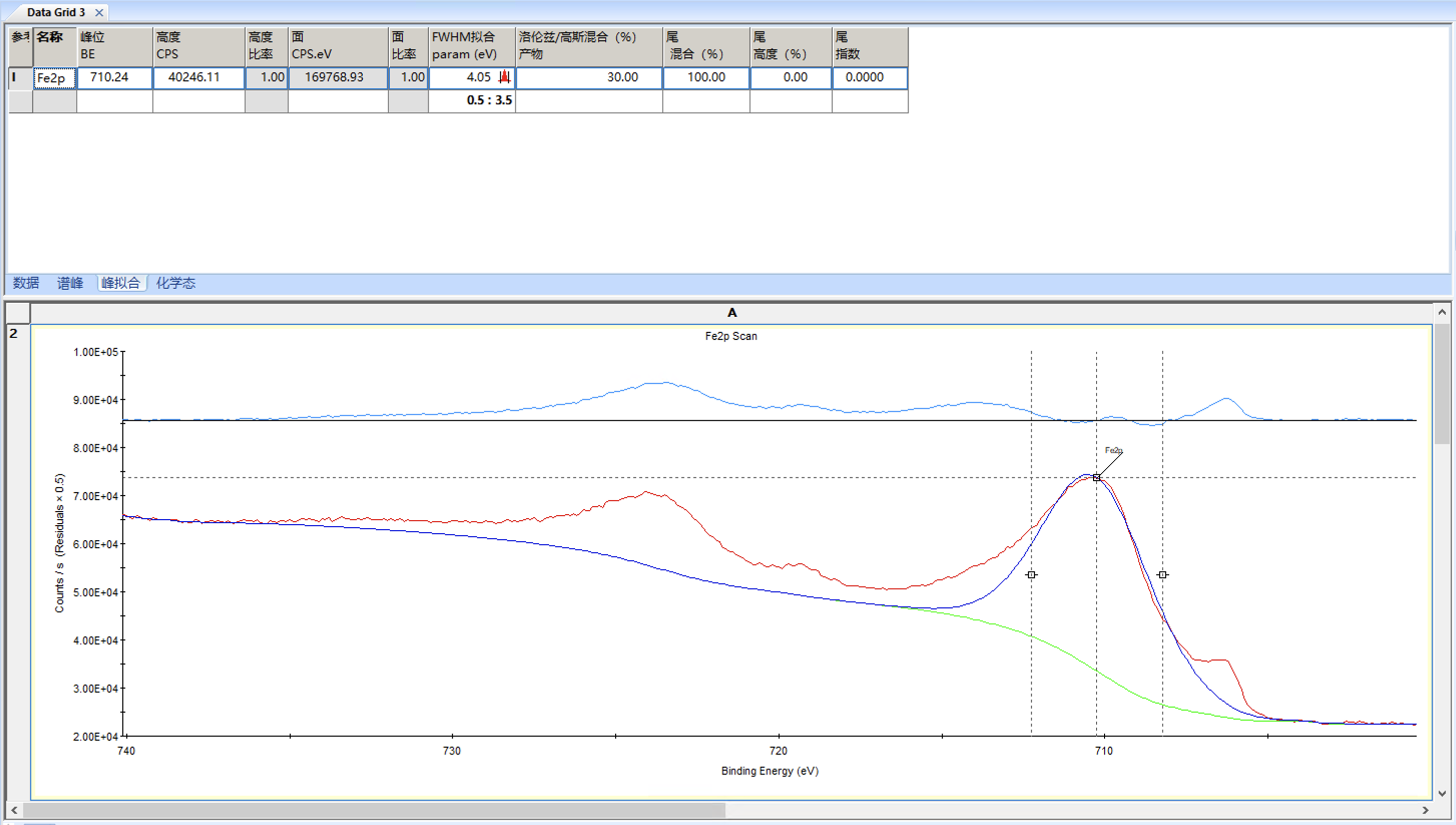
点击添加双峰
可以观察到为刚刚这个峰添加了其对应的2p 1/2峰
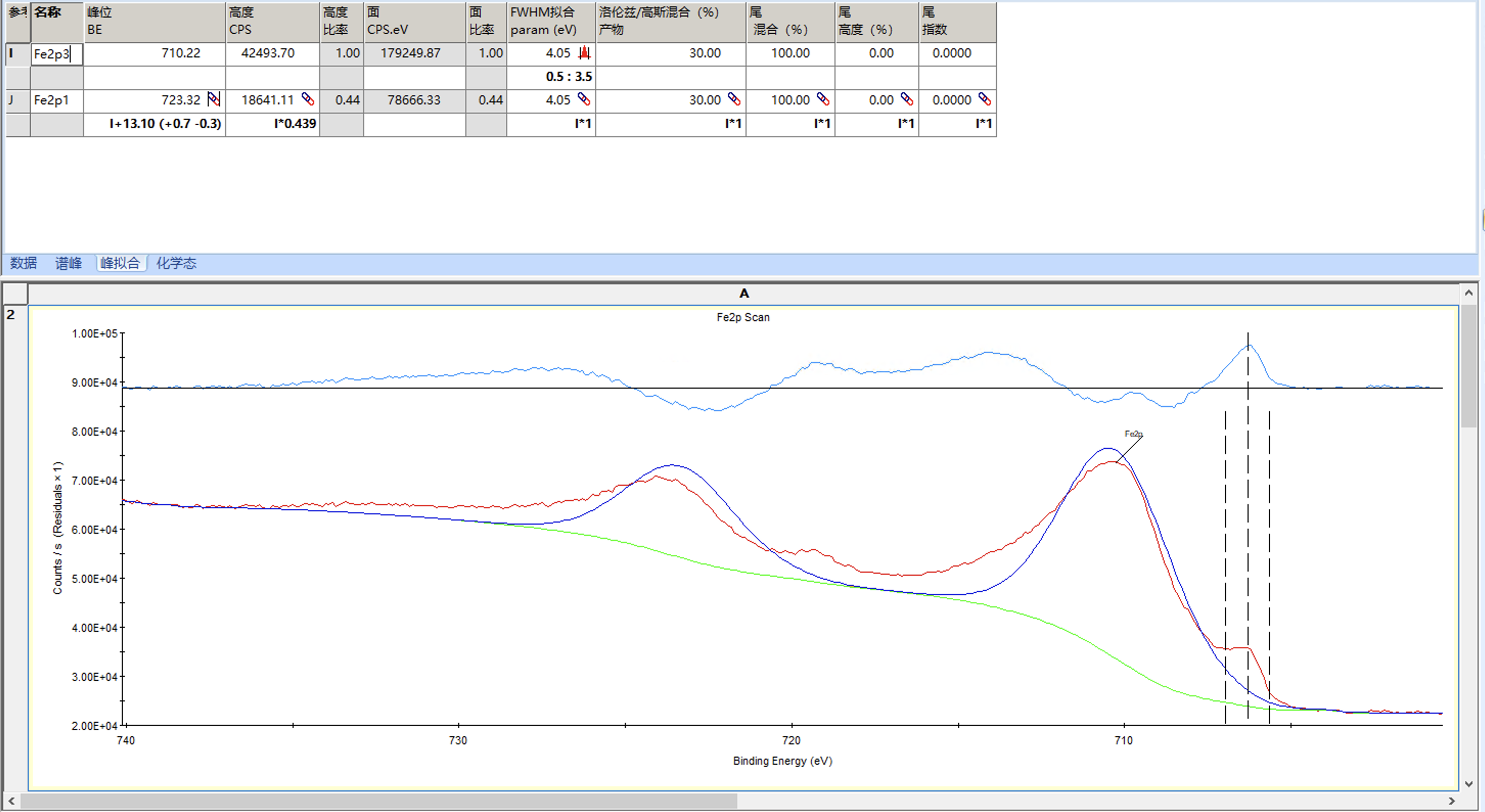
进行些许调整
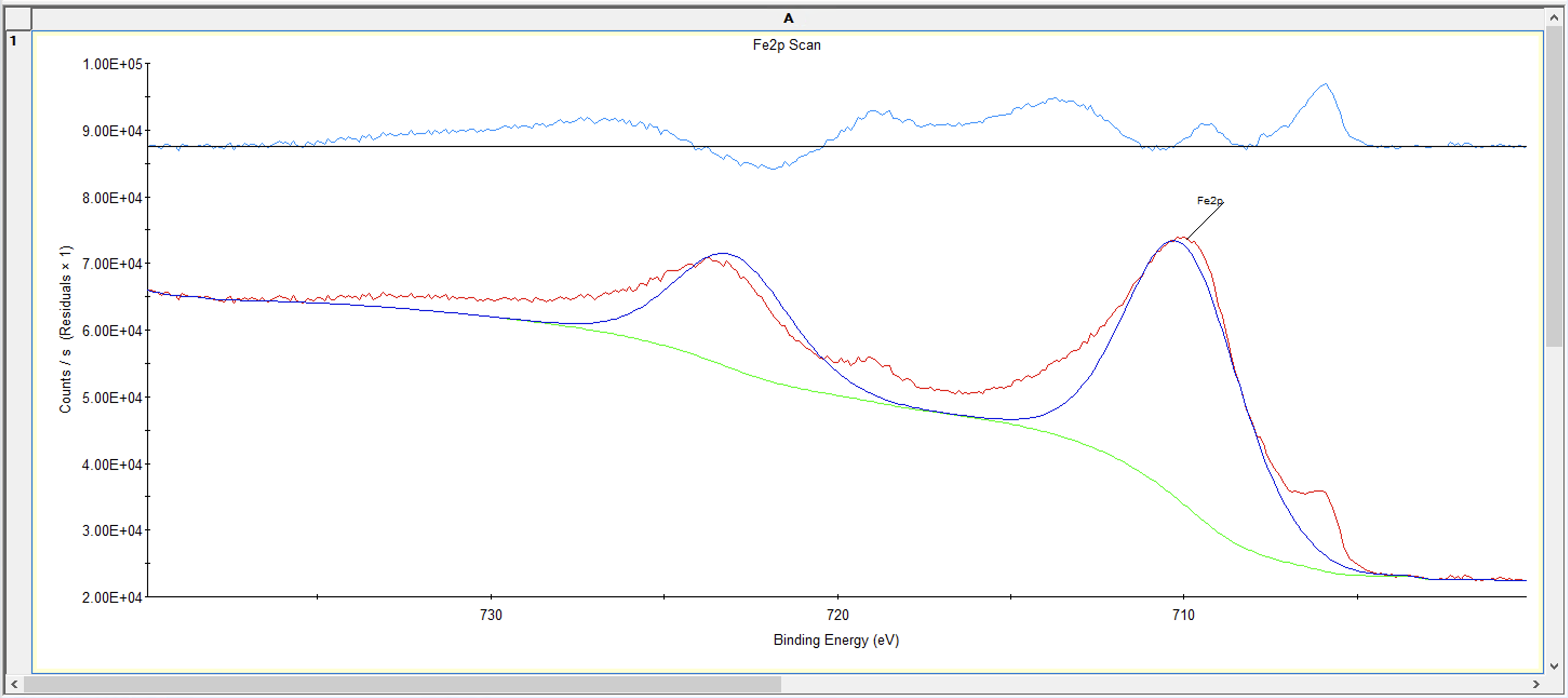
然后我们继续添加别的峰,我们的样品考虑有单质Fe、和2价Fe,而刚刚添加的是2价Fe,单质Fe大概在706.7eV附近,我们现在添加单质Fe的双峰
然后添加2价Fe的两个卫星峰,只需要分别添加单峰
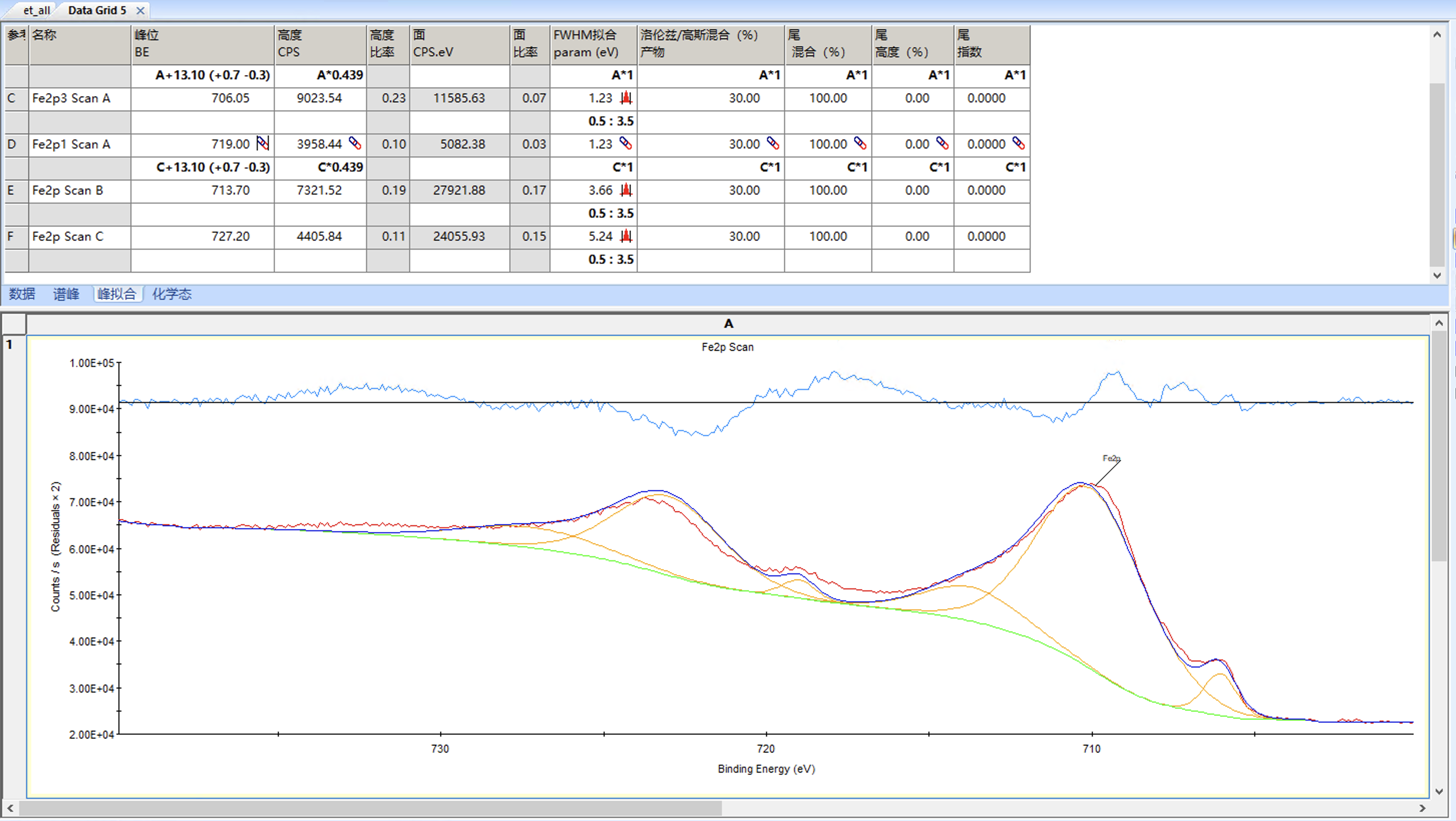
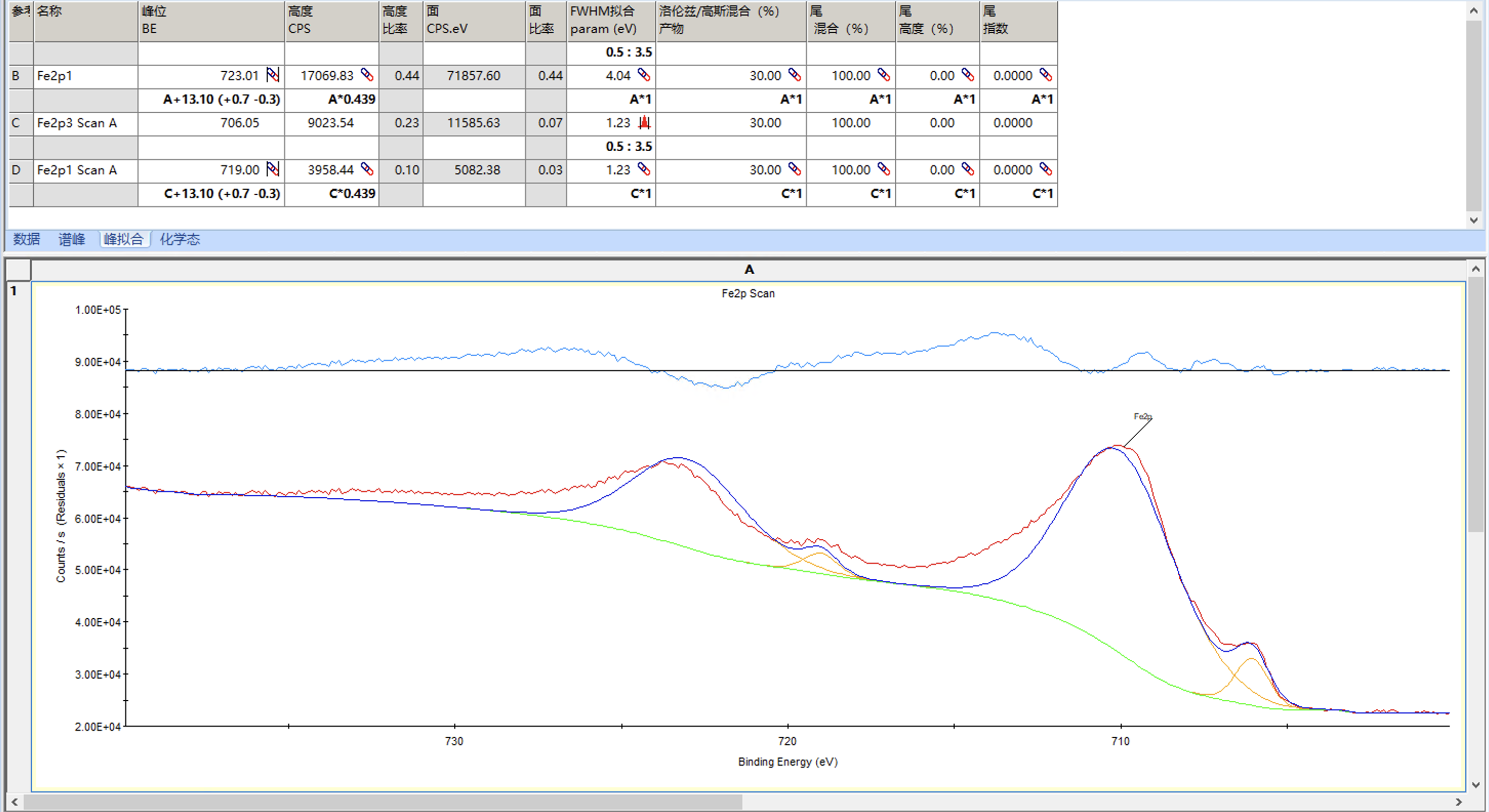
然后进行拟合,结果如下
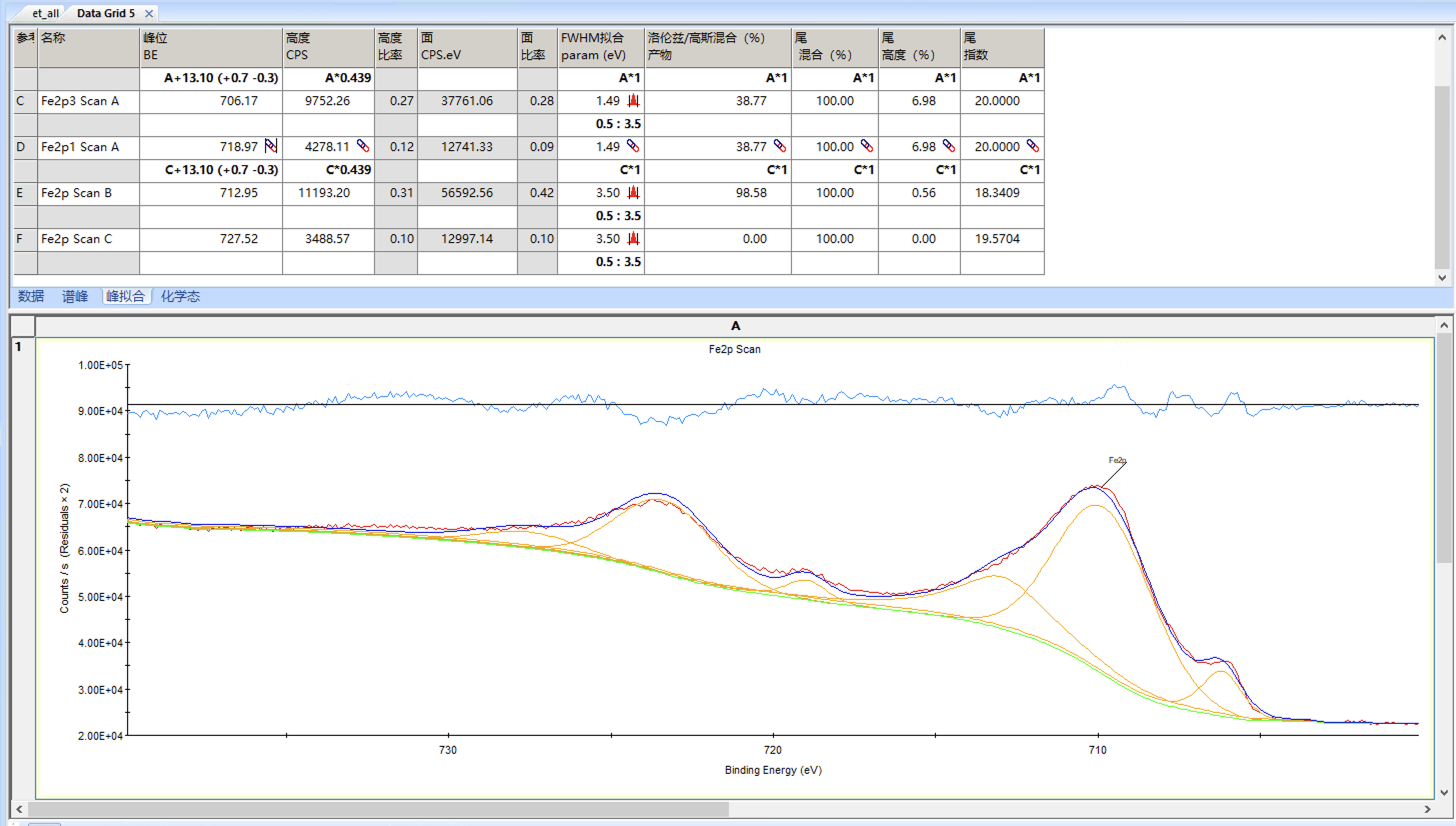
整体结果还不错,但是可以看到卫星峰的半峰宽已经达到量程3.5,而卫星峰的半峰宽通常会稍微大一些,我们可以适当增加量程重新拟合
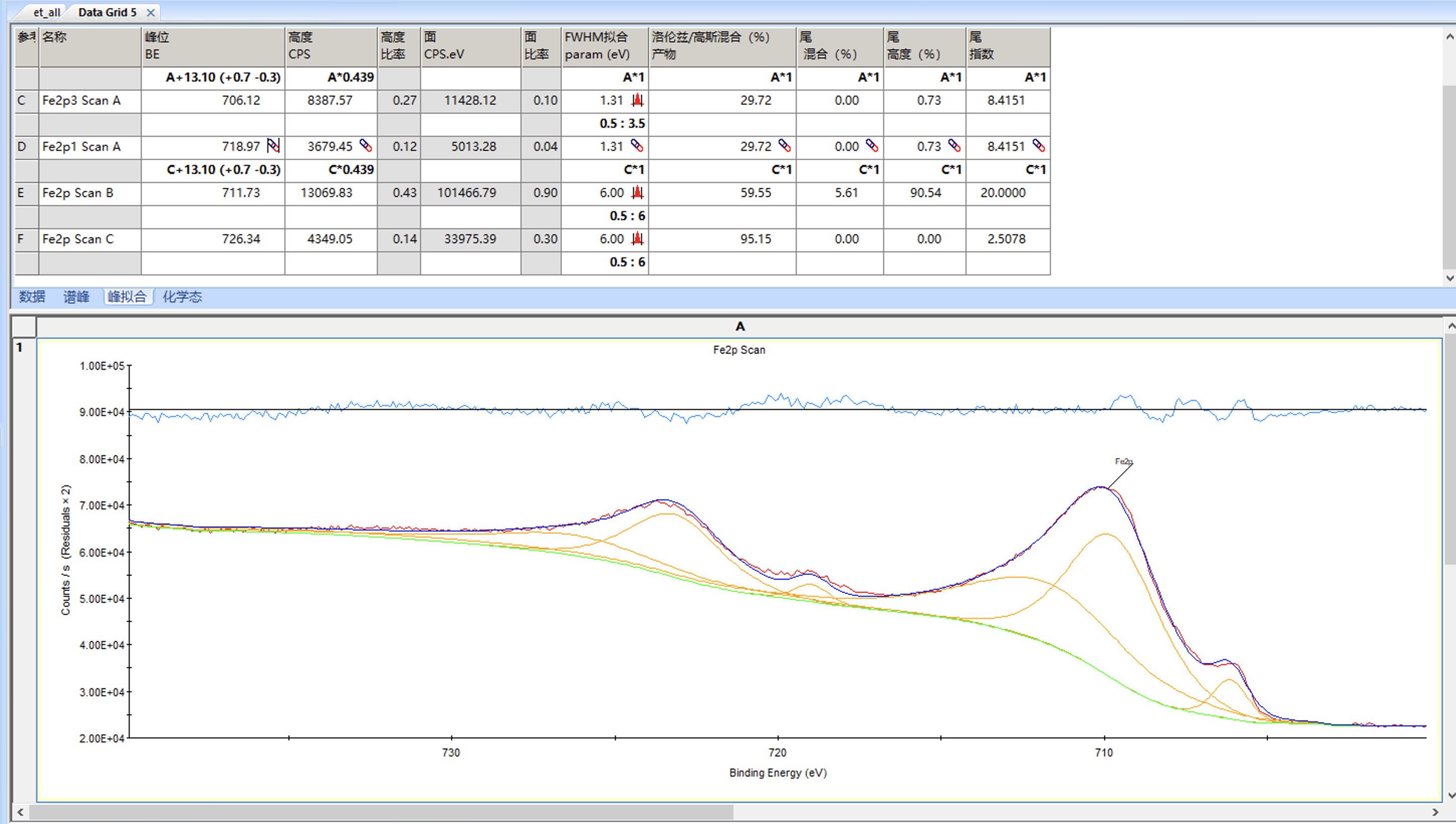
如果觉得没问题就可以接受结果,然后点确定就完成了
数据导出
从右上方的报告中打开报告选项
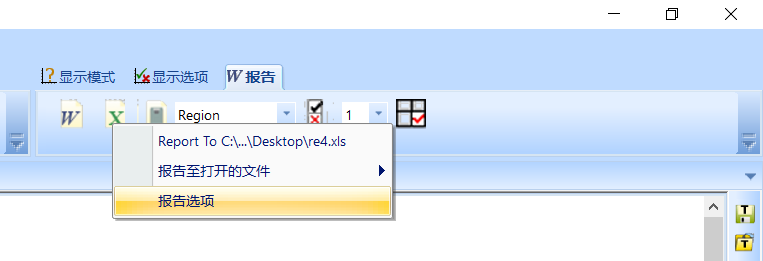
从表格选项中勾选峰位表
然后找到Excel的选项选择导出的路径和文件名后点击确定
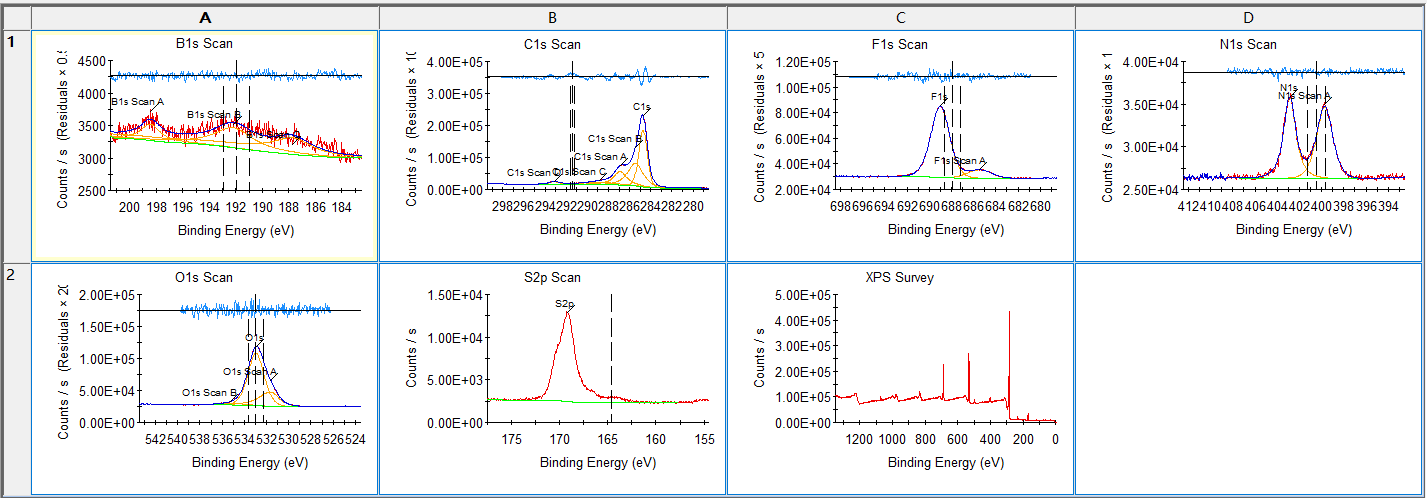
然后将所有谱图选中,或者选中要导出的谱图
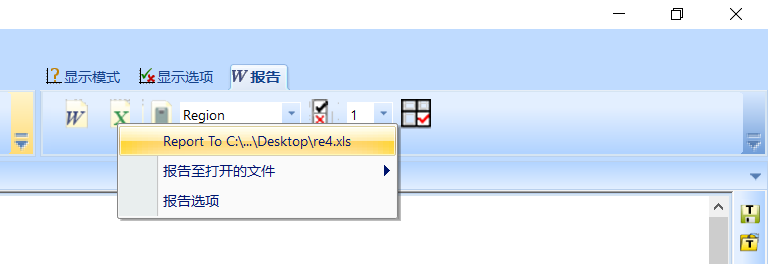
然后从右上角报告选项中选择Report To……,然后等待一会就完成了
PS:Avantage软件在校准页面经常卡死,原因是使用了中文输入法,保险起见,在校正之前要用英文输入法如下:

后记
原理没有特别深入的探讨,等以后有兴趣研究的话再更新吧,不过对于分析数据来说也够用了
最后,补充几个用于结合能分析的文件和查询网站,不过更推荐去论文中对应